First, log in to OOD: go to ood.bc.edu, then log in with your Andromeda credentials:
Once in, in the top panel, go to “Interactive Apps”, then “Gaussian View”:
After clicking on “Gaussian View”, set the account through which you will launch a GaussView (Gaussian) GUI, as well as the number of hours you want that session to run:
Once you have decided these, click “Launch”.
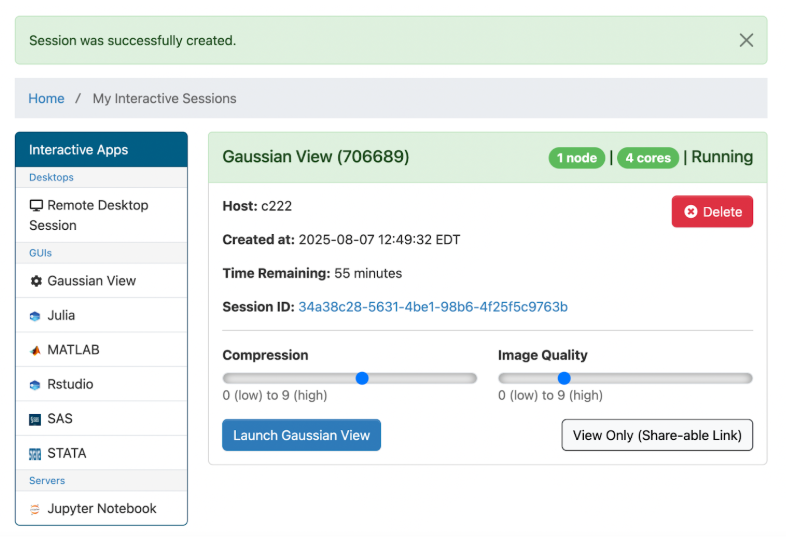
Click on “Launch Gaussian View”. You should be directed to a browser that looks like this (roughly; the panel by default will be at the top of the screen; I have moved it to be on the side of the screen for reasons of convenience):
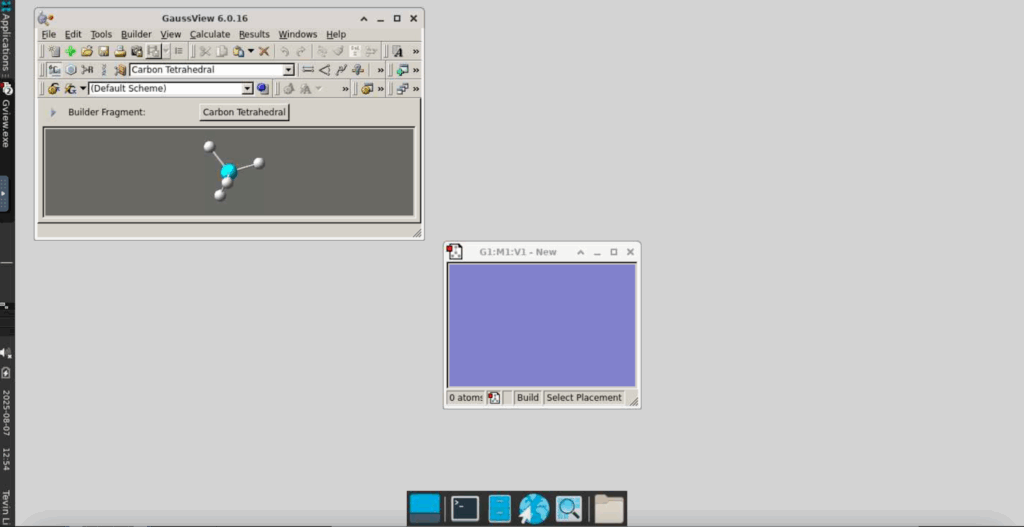
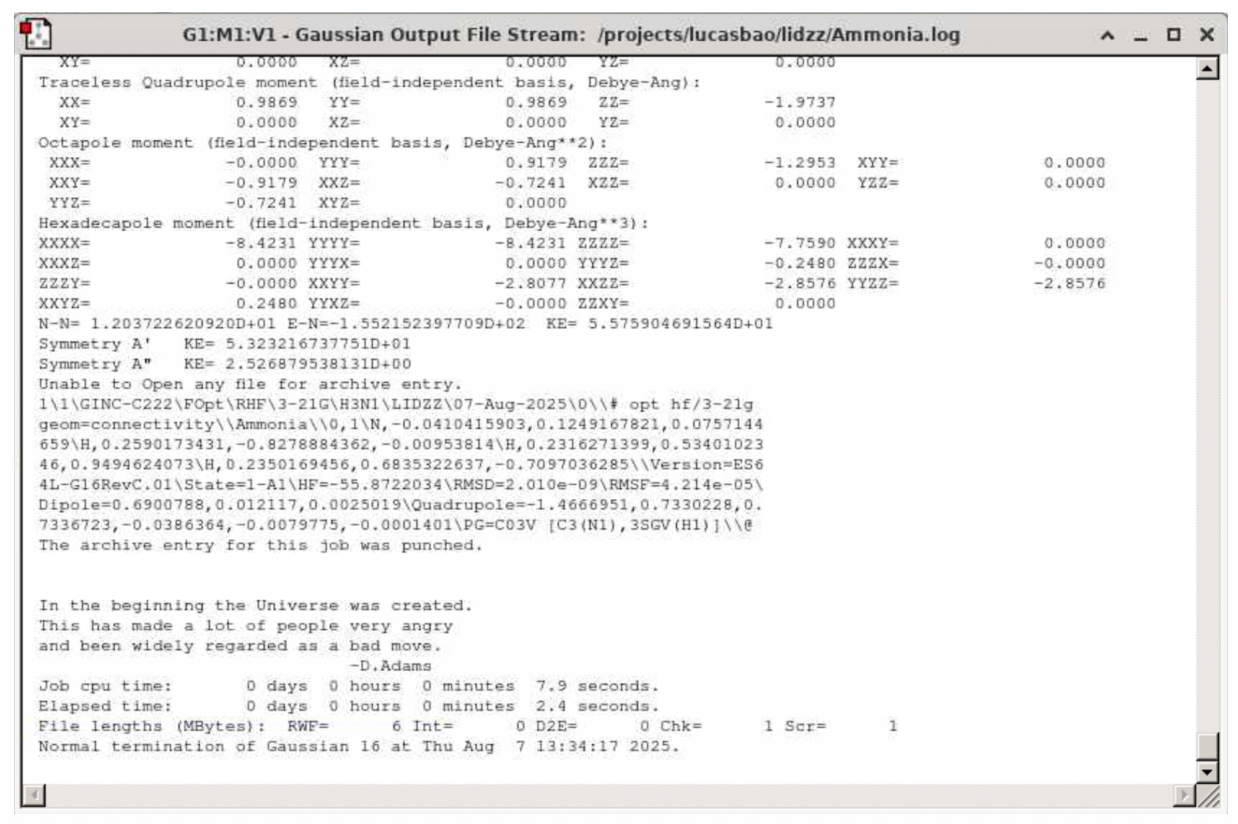
We will now begin a Gaussian optimization job, optimizing the structure of a simple NH3 (ammonia) molecule:
In the window labelled “GaussView 6.0.16”, click on “Carbon Tetrahedral”. This will lead you to a window like this:
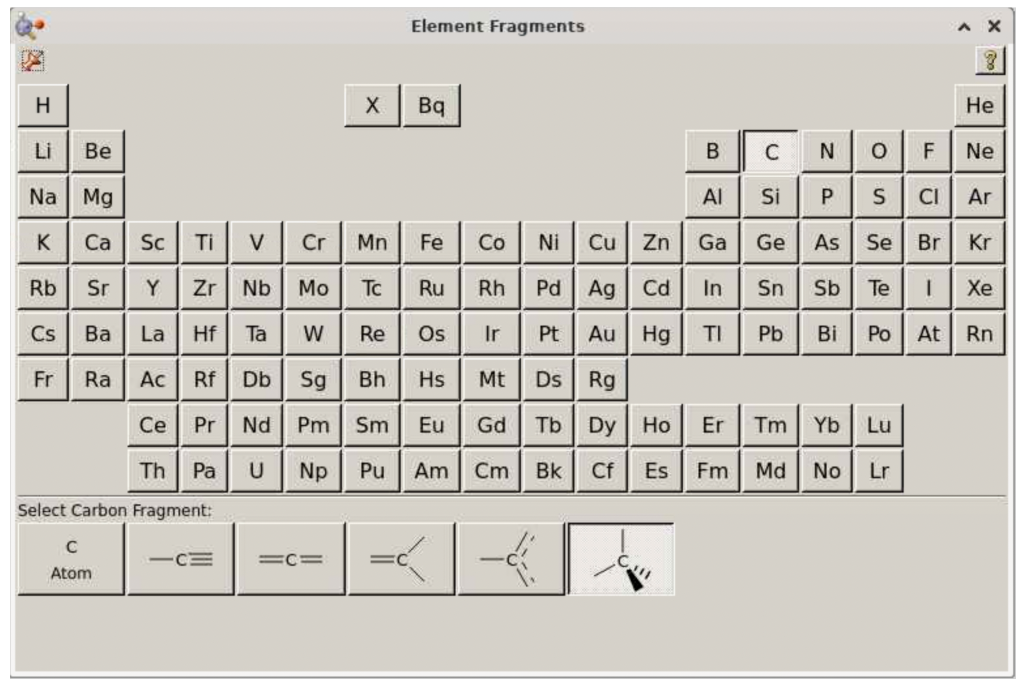
In the above window, select “N”: this should lead to this view:
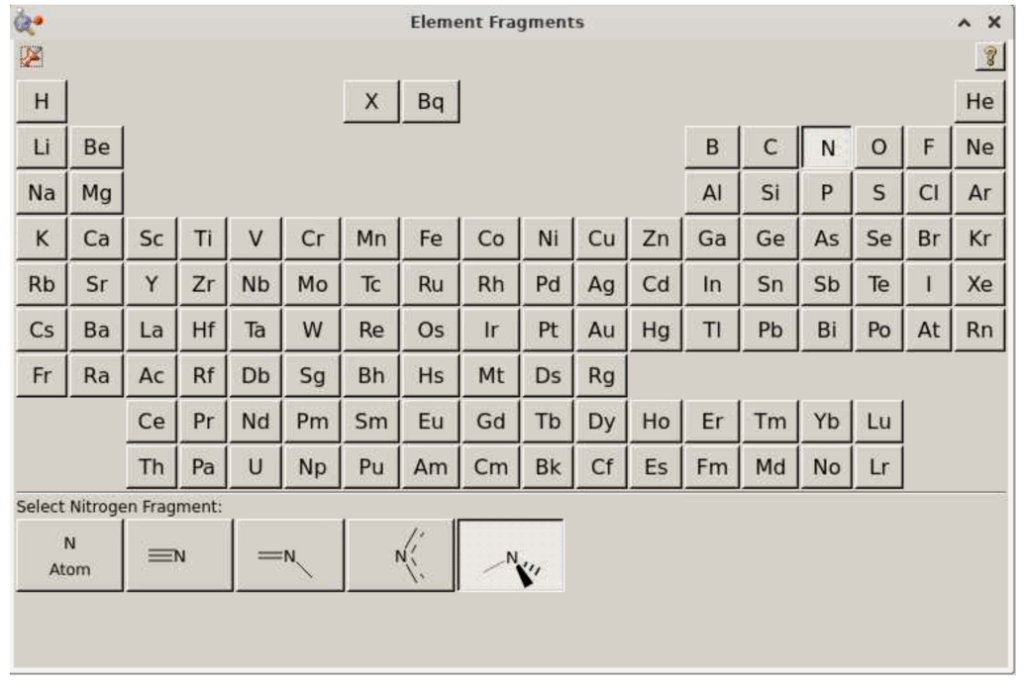
In the “Select Nitrogen Fragment” section, click on the rightmost option:

You should now see this on the screen:
Now, in the window that says “G1:M1:V1-New…”, simply click on any part of the blue screen and it will add the NH3 fragment to the screen (only click ONCE, otherwise, this will add multiple NH3 fragments):
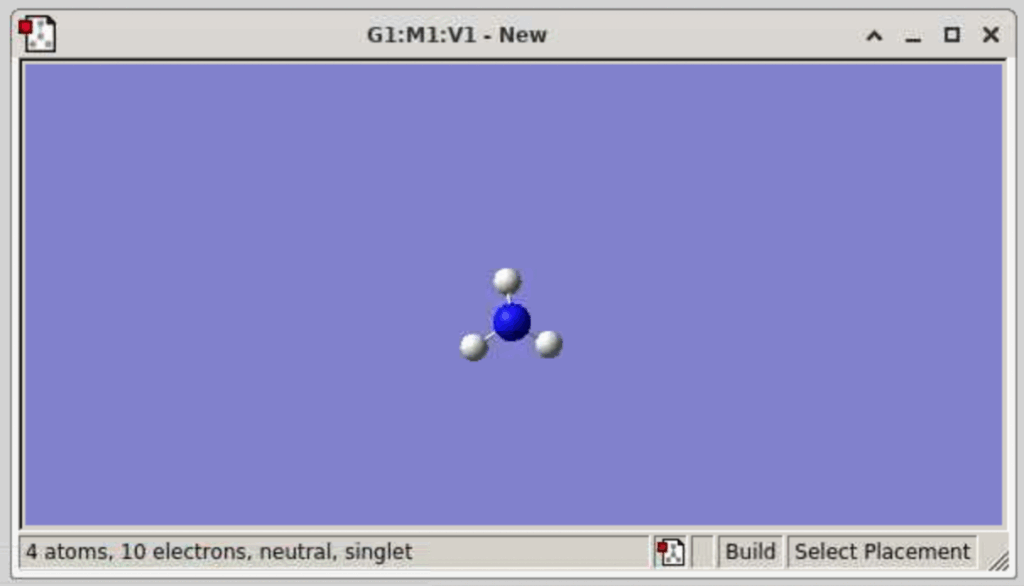
(If you want to rotate this molecule, click on any part of the molecule and drag it around. To move the molecule translationally, you will need to Control (or command) Shift then click on the molecule and drag it to wherever you want to on the blue screen. To Zoom in on/enlarge the molecule, you need to have control pressed down and then use your cursor to zoom in or out. You can also use your cursor to enlarge the window)
Now we want to set up a calculation for this molecule: in the “GaussView 6.0.16” window, select Calculate >> Gaussian Calculation Setup:
This will lead to the following window:
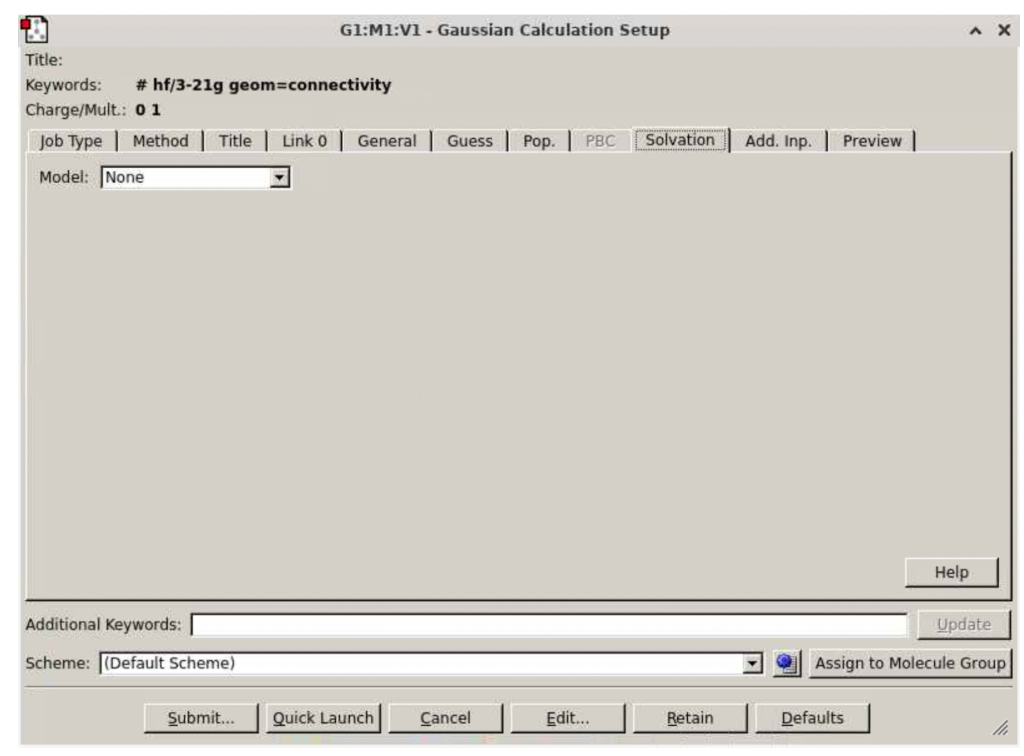
You can select any number of tabs to select whatever keywords you want for this particular run, according to the needs of your particular run. If you just want to follow the example below, perform the following instructions:
Under the Link0 tab, in “chkpoint” file, select “Specify”, then fill in “Ammonia.chk”.
In the “Job Title” tab, fill in “Ammonia”
In “Shared Processors”, select “4”
In “Memory limit”, select 100 MW.
In the “Preview” tab, check that your input looks like this:
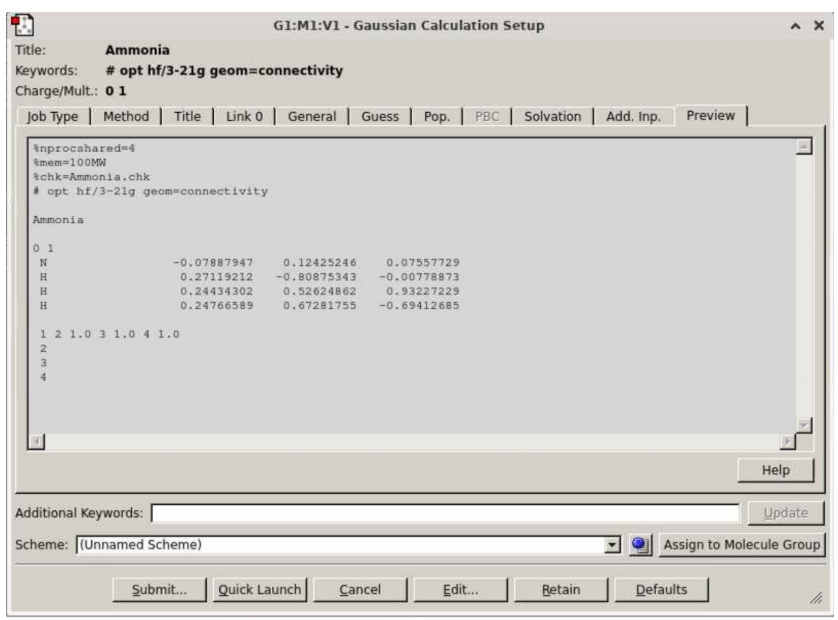
Click “submit”. It will prompt you to save the Gaussian input file (below): click Save
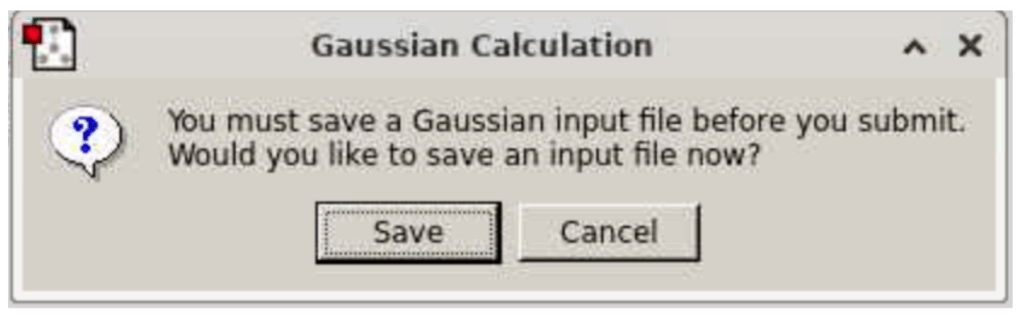
A window will appear where you can save your input file. Select a directory to store this input file, and, for file name, type “Ammonia” and, for “Files of Type”, select Gaussian Input files.
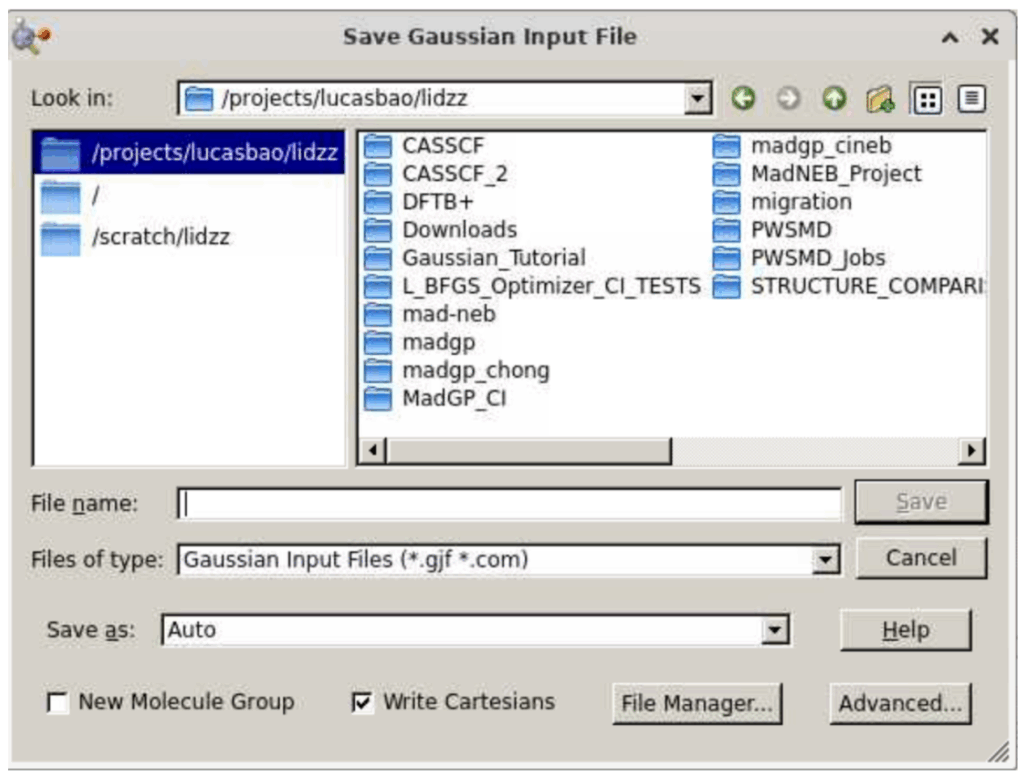
Then click save, at which point you will be directed to a window like this:
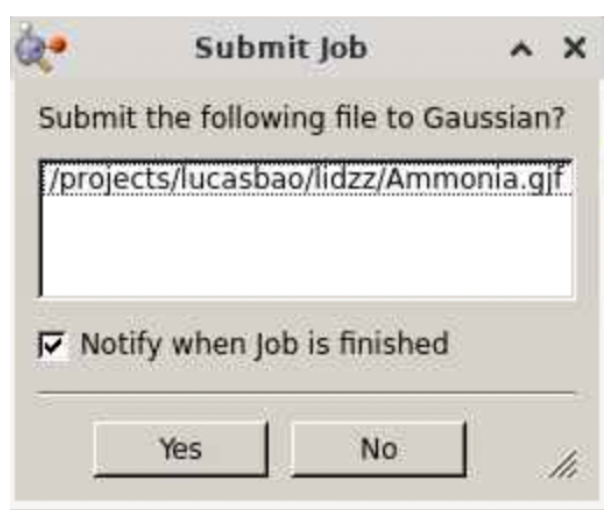
Select “Yes”. Your job will be submitted. (If your job is pending or not finished instantly, you can monitor its status by going to the Gaussview 6.0.16 window, selecting Calculate >> Current jobs, which will direct you to a job manager that shows all of your pending Gaussian calculations:
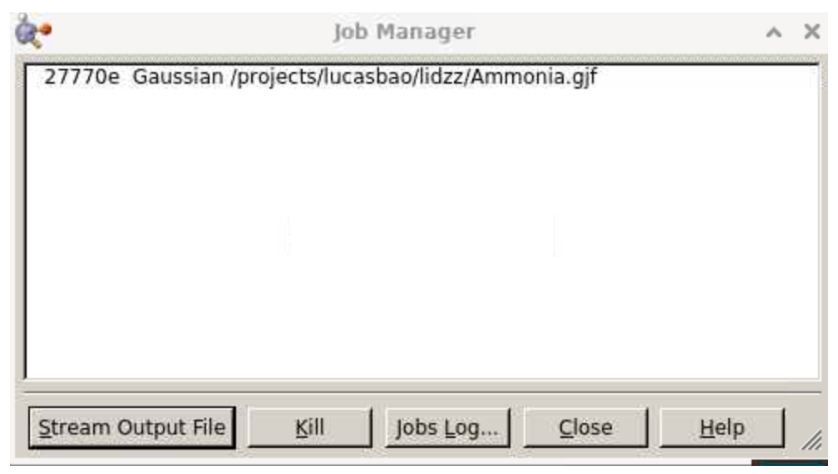
If you indeed selected “Notify when job is finished” above, you will receive a popup window like this when the calculation is done.
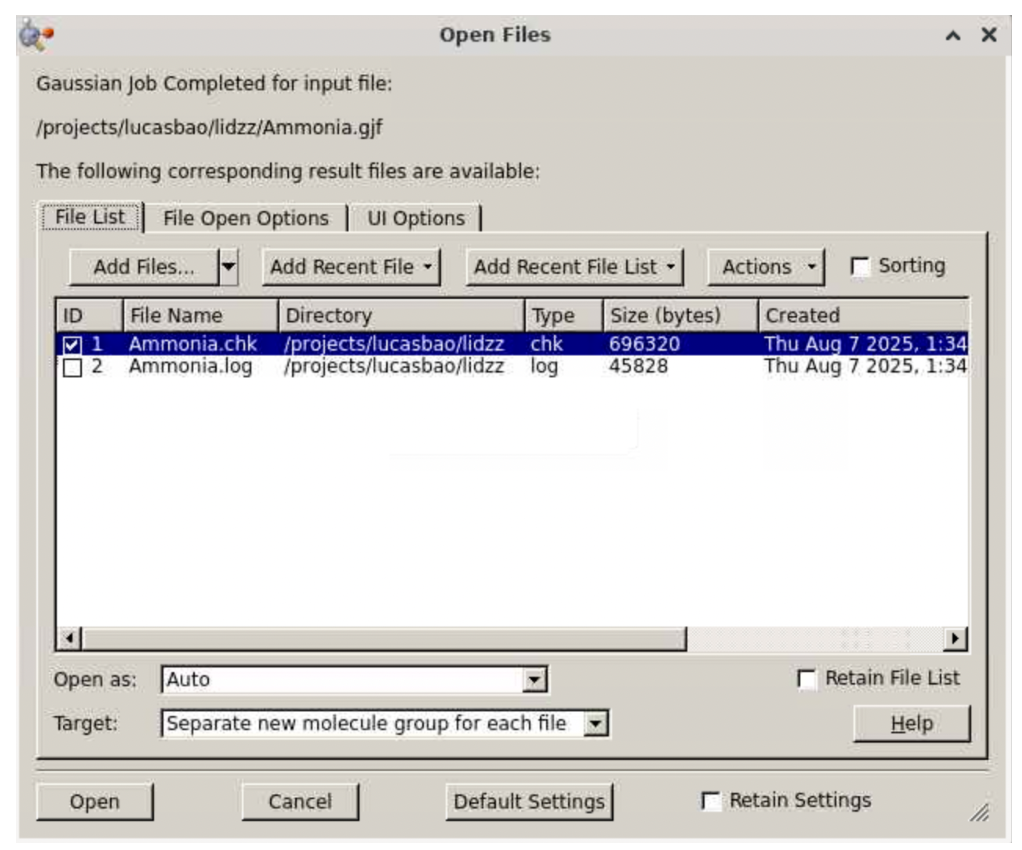
Check the file “Ammonia.log” (unchecking Ammonia.chk) and click “Open”. Verify that the job is indeed converged by going to the bottom of the file and searching for the keyword “Normal termination of Gaussian 16”: